Data Analysis¶
This section covers:
Exploring and summarizing data
Correlation and Chi-Squared tests
T-test and ANOVA
Checking assumptions
Linear regression
Set up¶
To get started let’s install and/or load the libraries we will be using. If this is your first time using one of the packages “uncomment” and run the appropriate install.package(‘package’)
#install.packages('tidyverse')
library(tidyverse)
#install.packages('car')
library(car)
#install.packages('broom')
library(broom)
#install.packages ('rstatix')
library (rstatix)
#install.packages("sjPlot")
library(sjPlot)
#install.packages("lmtest")
library(lmtest)
We are going to analyze penguins! See https://allisonhorst.github.io/palmerpenguins/
Let’s get the data
install.packages("palmerpenguins")
library(palmerpenguins)
Exploring the data set¶
View(penguins)
We can check the structure
str(penguins)
tibble [344 × 8] (S3: tbl_df/tbl/data.frame)
$ species : Factor w/ 3 levels "Adelie","Chinstrap",..: 1 1 1 1 1 1 1 1 1 1 ...
$ island : Factor w/ 3 levels "Biscoe","Dream",..: 3 3 3 3 3 3 3 3 3 3 ...
$ bill_length_mm : num [1:344] 39.1 39.5 40.3 NA 36.7 39.3 38.9 39.2 34.1 42 ...
$ bill_depth_mm : num [1:344] 18.7 17.4 18 NA 19.3 20.6 17.8 19.6 18.1 20.2 ...
$ flipper_length_mm: int [1:344] 181 186 195 NA 193 190 181 195 193 190 ...
$ body_mass_g : int [1:344] 3750 3800 3250 NA 3450 3650 3625 4675 3475 4250 ...
$ sex : Factor w/ 2 levels "female","male": 2 1 1 NA 1 2 1 2 NA NA ...
$ year : int [1:344] 2007 2007 2007 2007 2007 2007 2007 2007 2007 2007
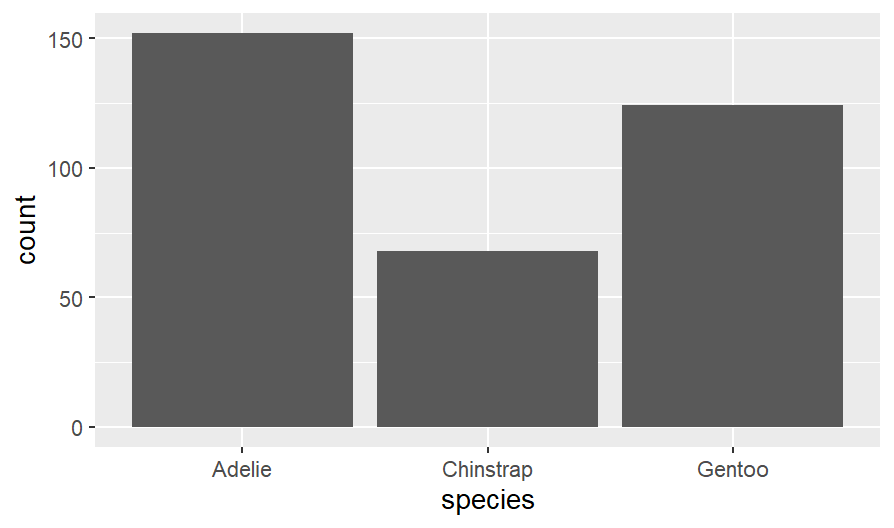
I want to see how many penguins I have
penguins %>%
count(species)
## # A tibble: 3 x 2
## species n
## <fctr> <int>
## Adelie 152
## Chinstrap 68
## Gentoo 124
## # 3 rows
Let’s create a bar graph
ggplot (penguins, aes(species))+
geom_bar()
I want to see summary statistics for each species of penguin
penguins %>%
group_by(species) %>%
summarize(across(bill_length_mm:body_mass_g, mean, na.rm = TRUE))
Correlation¶
Is there a correlation between Flipper Length and Body Mass? Let’s create a scatterplot first
correlation_graph <- ggplot(penguins, aes(x = flipper_length_mm, y = body_mass_g)) +
geom_point() +
geom_smooth(method = lm)
correlation_graph
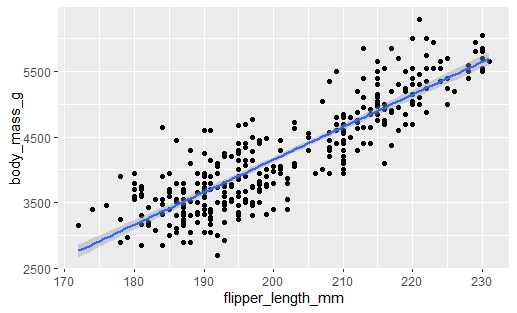
What’s the correlation coefficient?
cor.test(penguins$flipper_length_mm, penguins$body_mass_g)
## # Pearson's product-moment correlation
## data: penguins$flipper_length_mm and
## t = 32.722, df = 340, p-value <
## 2.2e-16
## alternative hypothesis: true correlation is
## not equal to 0
## 95 percent confidence interval:
## # 0.843041 0.894599
## sample estimates:
## # cor
## 0.8712018
Chi-Squared test¶
Now, I want to see if there is relationship between species and island. As both variables are categorical, we need to run a chi-squared test
Let’s visualize both varibles first
ggplot(penguins, aes(x = species, fill = island)) + geom_bar()
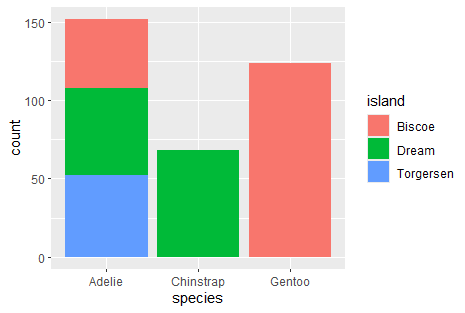
We can also build contigency tables
penguins_table <- table (penguins$species, penguins$island)
penguins_table
prop.table(penguins_table)
prop.table(penguins_table, 1)*100
prop.table(penguins_table, 2)*100
## # Biscoe Dream Torgersen
## Adelie 44 56 52
## Chinstrap 0 68 0
## Gentoo 124 0 0
## # Biscoe Dream Torgersen
## Adelie 0.1279070 0.1627907 0.1511628
## Chinstrap 0.0000000 0.1976744 0.0000000
## Gentoo 0.3604651 0.0000000 0.0000000
## # Biscoe Dream Torgersen
## Adelie 28.94737 36.84211 34.21053
## Chinstrap 0.00000 100.00000 0.00000
## Gentoo 100.00000 0.00000 0.00000
## # Biscoe Dream Torgersen
## Adelie 26.19048 45.16129 100.00000
## Chinstrap 0.00000 54.83871 0.00000
## Gentoo 73.80952 0.00000 0.00000
chi-squared test
chisq <- chisq.test(penguins$species, penguins$island)
chisq
## # Pearson's Chi-squared test
## data: penguins$species and penguins$island
## X-squared = 299.55, df = 4, p-value < 2.2e-16
Independent Samples t-test¶
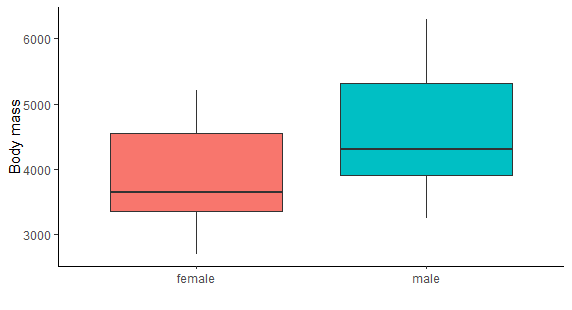
Is there a difference between males and females in their body mass?
Let’s visualize the data first
ggplot(na.omit(penguins))+
geom_boxplot(aes(x=sex, y=body_mass_g, fill=sex))+
theme_classic()+
ylab("Body mass")+
xlab('')+
theme(legend.position = 'none')
Are there any outliers? We can display these specific rows with the identify_outliers() function from the {rstatix} package
penguins %>%
group_by(sex)%>%
identify_outliers(body_mass_g)
## 0 rows | 1-4 of 10 columns
We can check to see if the two samples have equal variance by performing a leveneTest, inside the functions we put a formula of the structure ‘continuous_variable ~ grouping’.
leveneTest(body_mass_g~sex, data=penguins)
Levene’s test has a null hypothesis that the variances are equal, based on the results of our test we reject the null hypothesis.
## # Levene's Test for Homogeneity of Variance (center = median)
## Df F value Pr(>F)
##group 1 6.0586 0.01435 *
## 331
## Signif. codes:
## 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
We will use the t.test() to compare the two means, the default for the function is to assume variances are not equal, and it performs a Welch Two Sample t-test. If we have equal variance we have to set the argument var.equal=TRUE which would run a Two-sample t-test.
t.test(penguins$body_mass_g ~ penguins$sex)
## # Welch Two Sample t-test
## data: penguins$body_mass_g by penguins$sex
##t = -8.5545, df = 323.9, p-value =
##4.794e-16
## alternative hypothesis: true difference in means
## between group female and group male is not equal to 0
## 95 percent confidence interval:
## -840.5783 -526.2453
## sample estimates:
## mean in group female mean in group male
## 3862.273 4545.685
Effect Size. T-test conventional effect sizes: 0.2 (small effect), 0.5 (moderate effect) and 0.8 (large effect). For Welch t-test use ‘var.equal=FALSE’
penguins %>%
cohens_d(body_mass_g ~ sex, var.equal = TRUE)
## # A tibble: 1 x 7
## .y. group1 group2 effsize n1 n2 magnitude
## <chr> <chr> <chr> <dbl> <int> <int> <ord>
##1 body_mass_g female male -0.9369085 165 168 large
## # 1 row
ANOVA: Comparing means from multiple groups¶
I would like to see if there are differences in the bill length between species.
We can start with a visualization
ggplot(penguins, aes(x = species, y = bill_length_mm)) +
geom_boxplot()
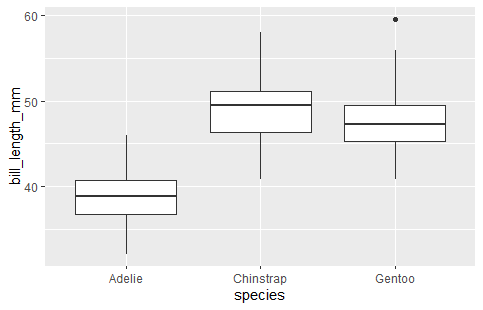
We can use the aov() function to run our anova. We will check our model assumptions after we fit the model. You want to assign the model fit to a variable name because we will use it to get the statistics and check assumptions.
anova<-aov(bill_length_mm~species, data=penguins)
summary(anova)
## # Df Sum Sq Mean Sq F value Pr(>F)
## species 2 7194 3597 410.6 <2e-16 ***
## Residuals 339 2970 9
## Signif. codes:
## 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
## 2 observations deleted due to missingness
Post-Hoc analysis to see where the differences are
Tukey_test<- TukeyHSD(anova)
Tukey_test
## # Tukey multiple comparisons of means
## 95% family-wise confidence level
## Fit: aov(formula = bill_length_mm ~ species, data = penguins)
## $species
## diff lwr upr p adj
## Chinstrap-Adelie 10.042433 9.024859 11.0600064 0.0000000
## Gentoo-Adelie 8.713487 7.867194 9.5597807 0.0000000
## Gentoo-Chinstrap -1.328945 -2.381868 -0.2760231 0.0088993
The tidyverse has a package called broom (we loaded this in earlier) and broom has a function called tidy() that we can use to create a cleaner table. We can also highlight significant differences with *
TukeyTidy<-TukeyHSD(anova)%>%
tidy()%>%
mutate(sig = case_when(adj.p.value < .05~ '*', TRUE ~''))
TukeyTidy
## # A tibble: 3 x 8
## term contrast null.value estimate conf.low conf.high adj.p.value sig
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <chr>
## species Chinstrap-Adelie 0 10.042433 9.024859 11.0600064 0.000000000 *
## species Gentoo-Adelie 0 8.713487 7.867194 9.5597807 0.000000000 *
## species Gentoo-Chinstrap 0 -1.328945 -2.381868 -0.2760231 0.008899333 *
## 1-1 of 3 rows
We can also get a plot .. tab:: R
par(mar = c(3, 8, 3, 3)) plot(Tukey_test, las=1)
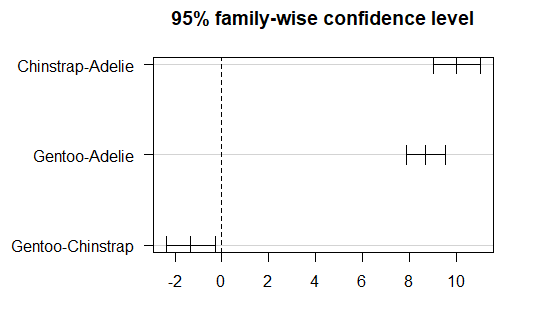
Checking the homogeneity of variance assumption
leveneTest(bill_length_mm ~ species, data=penguins)
## # Levene's Test for Homogeneity of Variance (center = median)
## Df F value Pr(>F)
## group 2 2.2425 0.1078
## # 339
Checking the normality assumption
plot(anova,2)
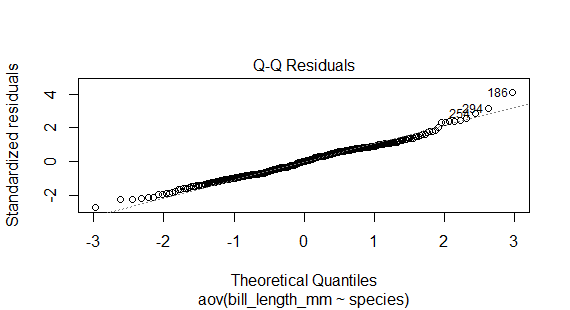
or we can use the Shapiro_Wilk test
anova_residuals <- residuals(object = anova )
shapiro.test(x=anova_residuals)
## # Shapiro-Wilk normality test
## data: anova_residuals
## W = 0.98903, p-value = 0.01131
The non-parametric alternative to one-way ANOVA is Kruskal-Wallis rank sum test, which can be used when ANNOVA assumptions are not met.
kruskal.test(bill_length_mm~species, data=penguins)
## # Kruskal-Wallis rank sum test
## data: bill_length_mm by species
## Kruskal-Wallis chi-squared = 244.14, df = 2, p-value <
## 2.2e-16
Linear Regression¶
A linear regression can be calculated the command lm. This function takes an R formula Y ~ X where Y is the dependent or outcome variable and X is the independent or predictor variable.
model1<-lm(body_mass_g ~ bill_length_mm, data=penguins)
summary(model1)
## # Call:
## lm(formula = body_mass_g ~ bill_length_mm, data = penguins)
## # Residuals:
## Min 1Q Median 3Q Max
## -1762.08 -446.98 32.59 462.31 1636.86
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 362.307 283.345 1.279 0.202
## bill_length_mm 87.415 6.402 13.654 <2e-16 ***
## ---
## Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
## Residual standard error: 645.4 on 340 degrees of freedom
## (2 observations deleted due to missingness)
## Multiple R-squared: 0.3542, Adjusted R-squared: 0.3523
## F-statistic: 186.4 on 1 and 340 DF, p-value: < 2.2e-16
We can add more variables
model2 <- lm(body_mass_g ~ bill_depth_mm + flipper_length_mm, penguins)
summary(model2)
## Call:
## lm(formula = body_mass_g ~ bill_depth_mm + flipper_length_mm,
## data = penguins)
## Residuals:
## Min 1Q Median 3Q Max
## -1029.78 -271.45 -23.58 245.15 1275.97
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) -6541.907 540.751 -12.098 <2e-16 ***
## bill_depth_mm 22.634 13.280 1.704 0.0892 .
## flipper_length_mm 51.541 1.865 27.635 <2e-16 ***
## ---
## Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
## Residual standard error: 393.2 on 339 degrees of freedom
## (2 observations deleted due to missingness)
## Multiple R-squared: 0.761, Adjusted R-squared: 0.7596
## F-statistic: 539.8 on 2 and 339 DF, p-value: < 2.2e-16
…and compare our models with the ‘tab_model’ function
tab_model(model1, model2, dv.labels = c("Model 1", "Model 2"))
Linear Regression assumptions
-Residuals are normally distributed. The plot()function will give us 4 diagnostic plots
res<-residuals(model2)
res <- as.data.frame(res)
ggplot(res,aes(res)) +
geom_histogram(fill='blue',alpha=0.5,bins=15)
plot(model2)
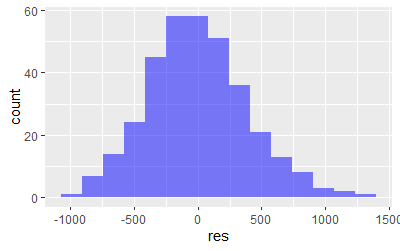
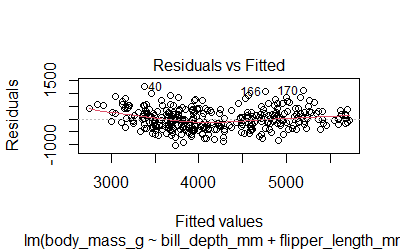
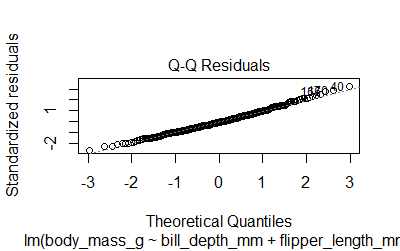
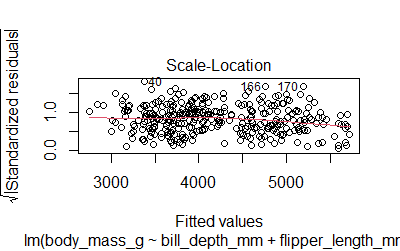
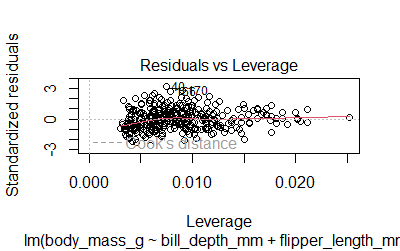
We can also run the Shapiro test
shapiro.test(model2$residuals)
## # Shapiro-Wilk normality test
## data: model2$residuals
## W = 0.99353, p-value = 0.1506
-Homoscedasticity. The variance of the residual in a regression model is constant. We can check the Scale-Location plot or use the Breusch-Pagan test (null hypothesis: heteroskedasticity is not present)
bptest(model2)
## # studentized Breusch-Pagan test
## data: model2
##BP = 2.4223, df = 2, p-value = 0.2979
-Multicollinearity. This issue occurs when two or more independent variables are highly correlated. We can use vif() to calculate the variance inflation factor. A value larger than 2.5 may be a cause for concern.
vif(model2)
## bill_depth_mm flipper_length_mm
## 1.51718 1.51718
Good resources to check out for more variations/details:
R cookbook by Paul Teetor
Learning Statistics with R by Danielle Navarro